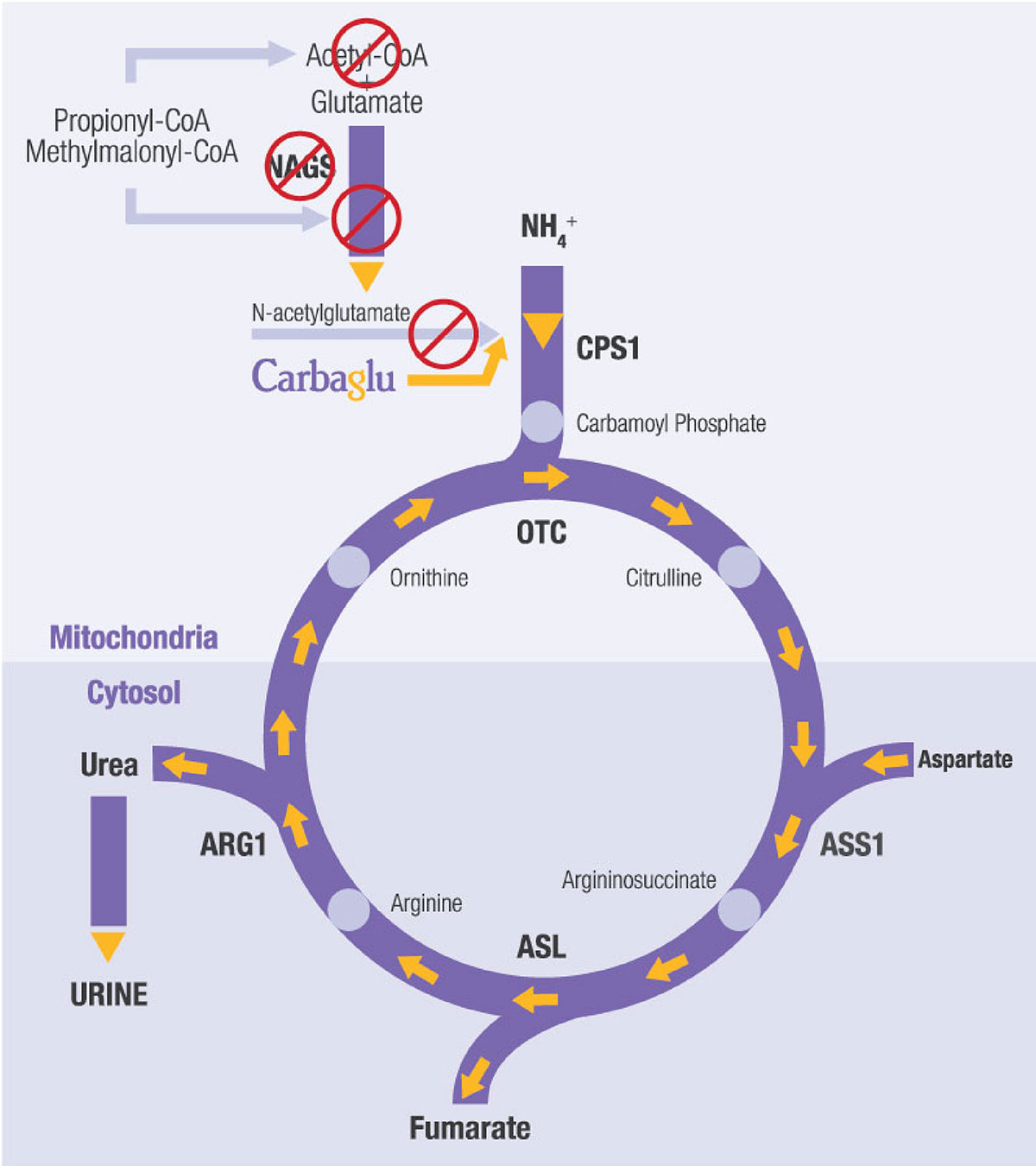
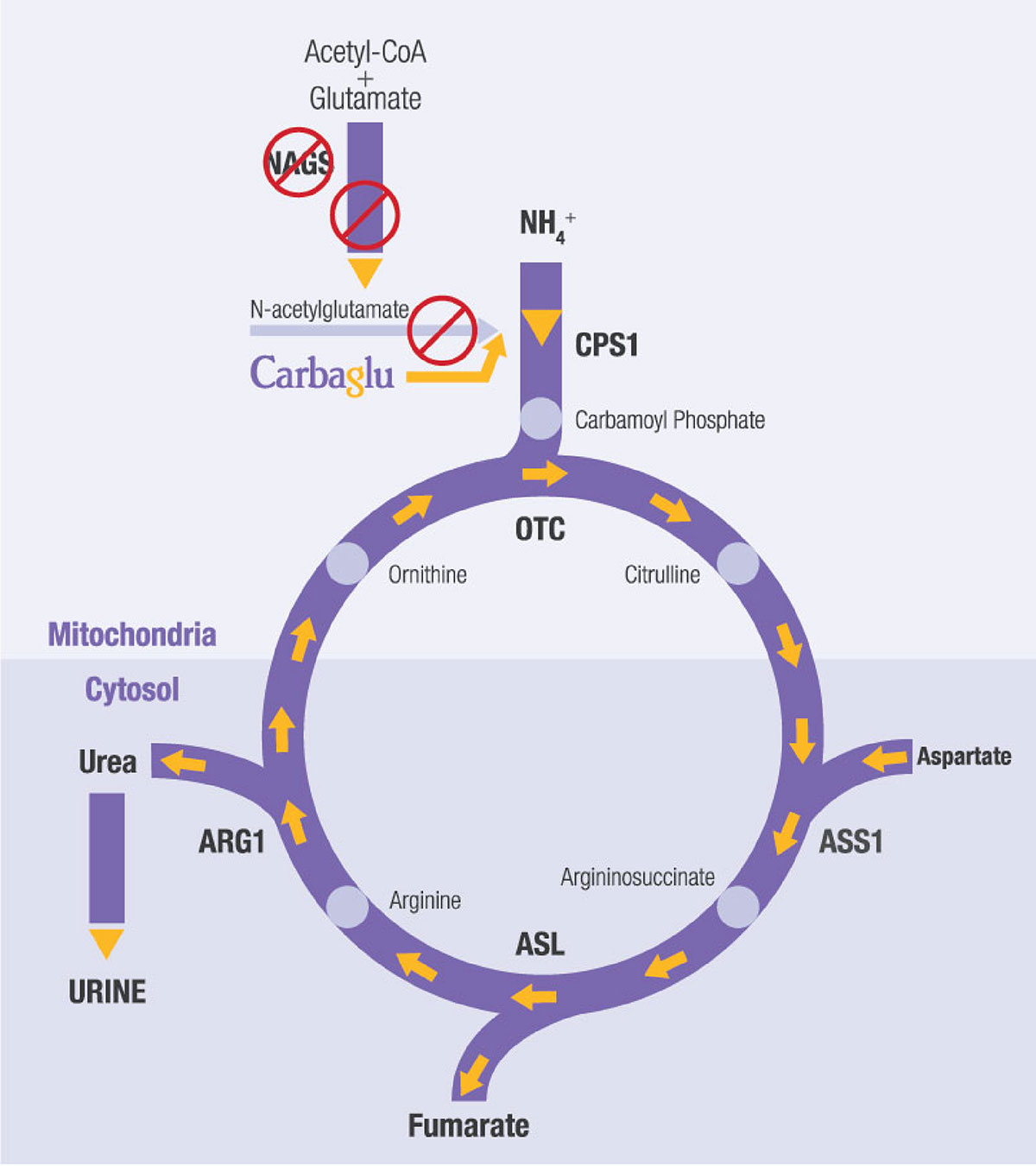
Mary has an extremely rare urea cycle disorder called NAGS (N-acetylglutamate synthase) deficiency. The disorder partially or entirely blocks the body’s natural process to eliminate excess ammonia, which leads to high ammonia build-up in the blood. This is dangerous because high blood ammonia can be extremely toxic to the brain. Mary’s journey to the right diagnosis would span 18 years, many hospitalizations, and thousands of miles.
“Babies are supposed to sleep”
Born in Belarus, Mary was a lethargic baby who tended to avoid foods containing protein as she grew. Mary’s mother, a physician, suspected something was wrong, but doctors “told her she was crazy,” says Mary. “They said, ‘Babies are supposed to sleep.'” Severe symptoms, including confusion and inability to focus, a “foggy brain” feeling, and irritability, led to a hospitalization when Mary was 10 years old. By then, the family had moved to the United States. A doctor checked Mary’s ammonia level, which was high, and a diagnosis was made: OTC (ornithine transcarbamylase) deficiency, a different urea cycle disorder than NAGS deficiency that can also cause high blood ammonia levels. Mary began taking a medication commonly prescribed for OTC deficiency. “It helped with some of my symptoms,” she recalled. But despite taking the medicine and eating a very low-protein diet, “I still ended up in the hospital pretty often.”
Added stress during her freshman year of college
Like many freshmen, Mary was excited about college, but her unpredictable symptoms caused anxiety. “I felt like I was always in crisis mode,” she says. “I knew I could feel fine one day and be in the hospital the next day.” One night after she and a friend had eaten at a restaurant, she vomited at a grocery store. By then she knew the drill. “I had to call my mom because I couldn’t even drive myself to the hospital,” she says. “I felt like my adulthood was being taken away from me.”
Mary recalls, “I was crying, thinking about how maybe the hospitalizations would continue, how my college studies and friendships would be affected.” She had only ever told her closest friends about her condition because it was hard to describe and she didn’t want it to define her.
A clinical trial: help for Mary at last
Neither Mary’s family nor her team of doctors had given up trying to find an answer to her continuing symptoms. And it was right before her final hospitalization during her freshman year that they found one, although it would be months before the answer could be verified and fully realized.
Mary’s doctors told the family about a treatment (now known as CARBAGLU® or carglumic acid) that was being investigated for urea cycle disorders including NAGS deficiency. Her doctors explained the potential risks and benefits of the medication. In Mary’s case, her doctors would be observing the response of someone with OTC deficiency to the medication. CARBAGLU treatment should be managed by a physician and medical team experienced in metabolic disorders.
Mary’s parents didn’t hesitate. “My mom said, ‘If it’s available, Mary’s going to do it.’ ” Of course, they didn’t know how Mary would react to CARBAGLU. During the study, her doctors noticed that her response to CARBAGLU was consistent with how a person with NAGS deficiency would respond. Later, Mary’s new diagnosis of NAGS deficiency was confirmed. After the trial, Mary waited anxiously for months for the Food and Drug Administration (FDA) to approve CARBAGLU for the treatment of NAGS deficiency so that she could resume treatment.
The correct diagnosis and treatment
Things changed after Mary was correctly diagnosed with NAGS deficiency and started taking CARBAGLU. “My life improved,” she says. It took a few weeks to safely decrease the medicine she had been taking and increase CARBAGLU to the appropriate dose. But then she was able to start eating a normal diet without any protein restrictions, meaning no more weighing foods and counting grams of protein.
While Mary remains aware of the possible risks of taking CARBAGLU, today her biggest worries revolve around making sure she always has it with her. “I keep it at my desk at work, just in case there’s a snowstorm and I can’t get home or I forget to take it one morning when I’m in a rush,” she says. She also packs some in every bag when she travels. “But you get into a habit. Now I just get up, take my medicine, go about my day, come back home, take my medicine, go to bed, and that’s it.”
A focus on the future
Mary’s care is overseen by a team of healthcare professionals that includes a metabolic specialist, a metabolic dietitian, and a genetic counselor. Because she’s medically stable, Mary sees the team just once a year. But they’re always available by phone if she has questions.
Mary’s doctors have told her and her family about foundations, conferences, events, research studies, and opportunities to connect with other people who have rare metabolic conditions. Being part of a community is very important to Mary. Although she’s still anxious about what could happen in the future, she focuses on what she can control – going to her doctor appointments, learning about her condition, and pursuing her goal of becoming a mental health therapist. “The right diagnosis made an incredible difference in my life,” she says.
Indications and Usage
Carbaglu® (carglumic acid) tablets for oral suspension 200mg is a prescription drug used in all ages to help treat the following:
• CARBAGLU is used to supplement standard of care treatment for acute high blood ammonia levels (hyperammonemia) due to the lack or shortage of NAGS enzyme activity.
• CARBAGLU is used to maintain normal blood ammonia levels due to the lack or shortage of NAGS enzyme activity.
• CARBAGLU is used to supplement standard of care treatment for acute high blood ammonia levels due to propionic acidemia (PA) or methylmalonic acidemia (MMA).
Important Safety Information
• Contraindications: None.
• NAGS deficiency: Most common side effects in ≥13% of patients are: vomiting, abdominal pain, fever, inflammation of the tonsils, reduced red cells in the blood, diarrhea, ear infection, infections, inflammation of the throat and nasal passages, hemoglobin decreased in red blood cells, and headache.
• PA and MMA: Most common side effects in ≥5% of patients are: lower than normal white blood cells, reduced red cells in the blood, vomiting, abnormal levels of minerals in the body, decreased appetite, low blood sugar levels, lack of energy/unresponsiveness, brain disease that alters brain structure or function, and inflammation of the pancreas/increase in a type of protein made by the pancreas that helps the body digest fats.
• To report SUSPECTED SIDE EFFECTS, contact Recordati Rare Diseases Inc. at 1-888-575-8344, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.