PA and MMA are disorders of propionate catabolism.1
PA is caused by a deficiency of the enzyme activity of propionyl-CoA carboxylase (PCC) resulting from mutations in either the PCCA or PCCB gene.1,8
MMA is most commonly caused by a deficiency of the enzyme activity of methylmalonyl-CoA mutase (MUT) resulting from mutations in the MUT gene. MMA may also be caused by defects in the transport or synthesis of adenosylcobalamin, a cofactor of MUT, resulting from mutations in the MMAA, MMAB, or MMADHC genes. These disorders are also known as cobalamin A (cblA) defect, cblB defect, and cblD-MMA defect, respectively.1,3
The deficiency of PCC enzyme activity in PA, or the deficiency of MUT enzyme activity or cofactor availability in MMA, impairs the body’s ability to completely catabolize the amino acids isoleucine, valine, methionine, and threonine; odd-chain fatty acids; cholesterol side chains; and propionic acid from the gut.1
As a result, toxic metabolites accumulate that can cause various biochemical abnormalities, including secondary hyperammonemia.6
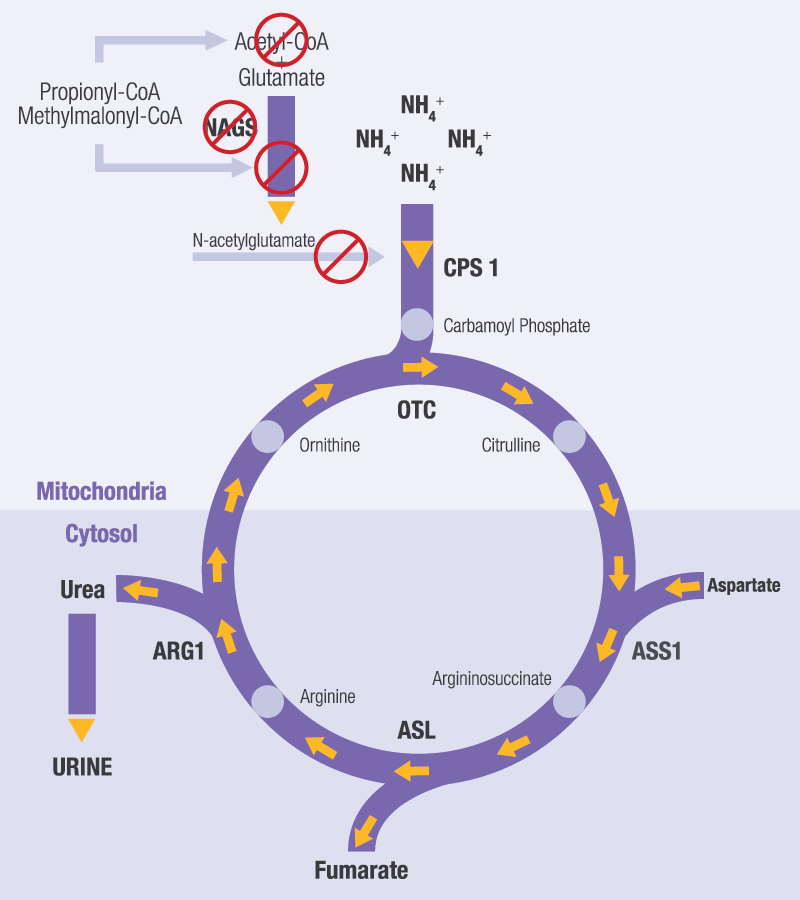
In PA and MMA, the urea cycle can become blocked due to:13,17
- Competitive inhibition of the enzyme N-acetylglutamate synthase (NAGS)
- Depletion of hepatic acetyl-CoA, so less is available to form N-acetylglutamate (NAG)
- Less NAG reduces activation of the enzyme carbamoyl phosphate synthetase 1 (CPS 1). NAG is a cofactor of CPS 1, which catalyzes the first reaction of the urea cycle.
This secondary impairment of the urea cycle can lead to acute hyperammonemia with neurotoxic effects during metabolic decompensation in PA or MMA.6
However, not all metabolic crises in PA or MMA will involve hyperammonemia. 8,9